
Abstract
Hybrid oncocytic/chromophobe tumor (HOCT) of the kidney represents a poorly understood clinicopathologic entity with pathologic features that overlap between benign renal oncocytoma (RO) and malignant chromophobe renal cell carcinoma (ChRCC). Consequently, characterization of HOCT and its separation from the foregoing entities are clinically important. The aim of this study was to describe the pathologic and molecular features of HOCT and to compare them with those of RO and ChRCC. We retrospectively identified a cohort of 73 cases with renal oncocytic tumors (19 RO, 27 HOCT, and 27 ChRCC) for whom clinical follow-up data were available by 2 tertiary care hospitals. All cases were sporadic except for 2 HOCTs that were associated with Birt–Hogg–Dubé syndrome. Lesional tissues were retrieved for molecular analysis. We performed targeted gene sequencing of all exons of 261 cancer related genes on a subset of HOCT samples (n = 16). Gene expression profiling of a customized codeset was conducted on 19 RO, 24 HOCT, and 27 ChRCC samples. Clinicopathologic characteristics as well as DNA copy number alterations, mutational and transcriptional features of HOCT derived from sequencing and expression profiling data are described and compared to those in RO and ChRCC. HOCTs were more frequently multifocal and did not exhibit mutations in genes that are recurrently mutated in RO or ChRCC but showed copy number alterations primarily involving losses in chromosomes 1 and X/Y. The mRNA transcript data show that HOCT can be separated from RO and ChRCC. Hence, HOCT appears to represent a distinct renal tumor entity with genomic features that are intermediate between those of RO and ChRCC.
Similar content being viewed by others
Introduction
Renal cell carcinoma subtypes are characterized by a suite of histologic, immunohistochemical, and molecular features. The subset of renal oncocytic neoplasms includes primarily three tumor types that exist on a clinicopathologic continuum, ranging from benign renal oncocytoma to indolent hybrid oncocytic/chromophobe renal tumor (HOCT) to the malignant chromophobe renal cell carcinoma [1,2,3,4]. By conventional pathologic examination, HOCTs harbor a mixture of cells with morphologic and immunophenotypic features that overlap with those of renal oncocytoma and chromophobe renal cell carcinoma [2, 5, 6]. Consequently, pathologists are reluctant to diagnose renal oncocytoma, particularly on the basis of core needle biopsy sampling, resulting in the non-specific and suboptimal biopsy diagnosis of “renal oncocytic neoplasm”, which is rendered in up to 11% of biopsied renal masses [7].
HOCTs were initially recognized in patients with Birt–Hogg–Dubé syndrome [8, 9], most of whom harbor mutations in the folliculin (FLCN) gene. Subsequently, sporadic cases of HOCT have also been reported, including in a patient with liver metastasis [10]. Very few cases of HOCT, however, have undergone molecular examination. Whereas renal oncocytoma and chromophobe renal cell carcinoma have been well characterized in terms of their DNA copy numbers, driver mutations, and transcriptomic profiles [11, 12], genomic examination of a series of histologically borderline HOCTs has been limited, and has yielded conflicting results: one group found multiple chromosomal gains and losses that were distinct from those in other renal cell carcinomas, including chromophobe renal cell carcinoma [13]; a second group found few copy number aberrations, similar to renal oncocytoma [14]. Moreover, profiling of driver mutations or transcripts has not been undertaken in HOCT series, underscoring the wide gaps in our molecular understanding of these tumors.
The aim of this study was to investigate where HOCTs cluster within the spectrum of renal oncocytic tumors at the clinicopathologic, copy number, mutational, and transcriptomic levels. We found that HOCTs may show aggressive behavior, lack classic renal cancer driver gene mutations and possess a molecular profile that is intermediate between renal oncocytoma and chromophobe renal cell carcinoma. Our findings justify considering HOCTs as distinctive renal oncocytic tumors of low malignant potential.
Materials and methods
Patient and tumor characteristics
For this retrospective study, we searched our institution’s database for surgically resected kidney cases using the keywords “hybrid”, “oncocytic”, “oncocytoma” and “chromophobe” between 2003 and 2015. We found 328 renal oncocytomas, 271 chromophobe renal cell carcinomas (including 24 cases with metastasis or sarcomatoid features), 126 renal oncocytic neoplasms, 87 clear cell or papillary renal cell carcinomas with oncocytic features, and 15 cases with the term “hybrid” in the final diagnostic line. Review of the pathology reports yielded 68 cases with equivocal diagnoses: among these, we found 25 cases where three genitourinary pathologists (PR, PT, KS) agreed on a consensus diagnosis of HOCT after histologic review. In 43 cases, other diagnoses were favored, including renal oncocytoma (n = 29); eosinophilic variant of chromophobe renal cell carcinoma (n = 7); oncocytic papillary renal cell carcinoma (n = 2); clear cell renal cell carcinoma (n = 1); unclassified renal cell carcinoma (n = 4). In addition, two cases with equivocal features of renal oncocytoma and chromophobe renal cell carcinoma from McGill University were sent to us, re-reviewed by all pathologists who agreed to place them in the HOCT category. The examined cohort comprised a total of 27 HOCT, 19 renal oncocytoma, and 27 chromophobe renal cell carcinoma cases. Paired non-neoplastic kidney tissues were used as controls. Diagnoses were confirmed based on hematoxylin- and eosin-stained sections and immunohistochemical stains performed as part of the initial workup of the case, when available. Additional histochemical or immunohistochemical stains were not performed as part of the study for case selection. The criteria used for diagnosis of HOCT included ambiguous architectural and cytologic features such as nuclear membrane irregularities and perinuclear clearing with oncocytic cytoplasm that could not be confidently placed into either renal oncocytoma or chromophobe renal cell carcinoma categories. Archived formalin-fixed, paraffin-embedded tissues and frozen samples from tissue banks at The University of Texas MD Anderson Cancer Center (Houston, TX) and McGill University (Montreal, QC) were obtained after informed consent and using an institutional review board-approved protocol (IRB# LAB 08–670). Lesional tumor foci as well as non-neoplastic kidney controls were marked on hematoxylin- and eosin-stained slides from all cases. These foci were manually macrodissected with a blade prior to the extraction of nucleic acids. For HOCTs, since the vast majority of cases showed composite morphologic features rather than clearly defined oncocytoma-like and chromophobe-like zones, microdissection of separate areas was not feasible and was not performed. The clinicopathologic features of the study cohort and survival data are summarized in Table 1. Detailed information on all cases is presented in Supplementary Table 1.
Whole-exome sequencing
We performed whole-exome sequencing in a selected group of patients with suspicion of Birt–Hogg–Dubé syndrome, including younger age, multifocal lesions, and renal oncocytosis to exclude mutations in the FLCN gene. Upon quantification and quality assessment of genomic DNA, the DNA from each sample (100–500 ng of genomic DNA) was sheared by sonication and libraries were prepared for sequencing following a standard laboratory protocol (Supplementary Methods). The captured libraries were sequenced on a HiSeq 2000 platform (Illumina Inc., San Diego, CA, USA) on a version 3 TruSeq paired end flow cell according to manufacturer’s instructions. We called single nucleotide variants and small indels using an in-house developed analysis pipeline. We then examined the coding regions in the FLCN gene to look for mutations. Detailed information can be found in the Supplementary Methods.
Targeted exome sequencing and copy number alterations
Genomic DNA was extracted using the QIAamp DNA Mini kit (Qiagen) according to the manufacturer’s instructions. The DNA concentration was individually quantified using a NanoDrop 1000 spectrophotometer (NanoDrop Technologies) and the Quant-iT Picogreen kit (Life Technologies). Next-generation sequencing involved target capture and sequencing of all exons of 261 cancer-associated genes using an in-house-developed and validated assay (T200.1), as previously described [15]. Details are provided in the Supplementary Methods with the list of assayed genes in Supplementary Table 4.
RNA extraction
Total RNA was extracted using the RNeasy FFPE extraction kit (QIAGEN, cat#73504) according to the manufacturer’s instructions. RNA was quantified by NanoDrop 2000, and its quality was assessed by an Agilent Bioanalyzer 1000 with an Agilent RNA 6000 Nano Kit (Agilent, cat#5067–1511). Details are provided in the Supplementary Methods.
Differentially expressed gene selection and NanoString nCounter gene expression assay
We initially performed an in silico analysis of publicly available gene expression data [12, 16, 17] to nominate the most differentially expressed genes between renal oncocytoma and chromophobe renal cell carcinoma and genes associated with aggressive chromophobe renal cell carcinoma [11, 18]. We selected genes that were the most differentially expressed in terms of fold change, as well as those genes that were found to be differentially expressed across multiple studies. We then evaluated the foregoing expression signature of renal oncocytic neoplasms on formalin-fixed paraffin-embedded tissues using Nanostring’s nCounter (NanoString Technologies) platform, a more feasible option for assaying clinical samples. Probe sequences were custom designed for target genes and manufactured by NanoString Technologies (Seattle, WA). This codeset included 100 target genes and 14 housekeeping genes. The housekeeping genes were selected from publicly available databases on the basis of their stability and detectable expression levels across the tissue type of interest, which were used to correct for RNA input amount or quality differences. The differential genes and housekeeping genes are listed in Supplementary Table 2. Manufacturer’s protocols were followed to perform the assay. Details are provided in the Supplementary Methods.
Statistical analysis
Additional statistical tests were used to analyze clinical and genomics data, including the Chi-square test, Fisher’s exact test, Mann–Whitney Wilcoxon test, and Cox proportional hazard analysis. Significance was defined as P < 0.05.
Results
Clinical and pathologic characteristics of sporadic HOCTs differ from those of renal oncocytoma and chromophobe renal cell carcinoma
We assessed 27 HOCTs with a median tumor diameter of 4.1 cm from a cohort whose median age was 63 years (range: 28–82 years) and 66% were men. Two patients showed stigmata of Birt–Hogg–Dubé syndrome in the form of fibrofolliculomas of the skin and 4 patients had kidneys with a background of oncocytosis (including the aforementioned 2 patients with Birt–Hogg–Dubé syndrome) as shown in Table 2. As illustrated in Supplementary Table 1, the right and left kidneys were involved in 12/27 (44%) and 11/27 (41%) cases, respectively; bilateral involvement occurred in 4/27 (15%) patients. Notably, one case was derived from a liver metastasis. The large majority of patients (70%, n = 19) with HOCTs harbored solitary renal masses; however, bilateral and multifocal tumors occurred in 15% (n = 4) and 30% (n = 8) of patients, respectively. Among sporadic HOCTs, multifocal presentation was seen in 24% (6/25) of patients. By contrast, renal oncocytoma and chromophobe renal cell carcinoma were multifocal in 11% (2/19) and 11% (3/27) of cases, respectively. Among the multifocal chromophobe renal cell carcinoma cases, however, only one case showed another tumor focus with chromophobe renal cell carcinoma histology whereas the other two cases showed clear cell renal cell carcinoma histology in the separate tumor focus (Supplementary Table 1). Two cases of HOCT showed aggressive clinicopathologic features: a liver metastasis resulting in cancer related death and a case with a renal cell carcinoma of unclassified type, ISUP/WHO grade 4, arising within a background of HOCT. Both of these cases were sporadic and confirmed to not represent another renal cell carcinoma subtype (eg., SDHB associated renal oncocytoma or MiTF Renal cell carcinoma) based on retained immunohistochemical expression of SDHB and immunonegativity for Cathepsin K, HMB45, and TFE3.
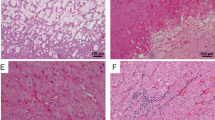
HOCTs were ambiguous in that they did not show unequivocal morphologic features of either renal oncocytoma or chromophobe renal cell carcinoma. Representative macroscopic and microscopic features of HOCTs are illustrated in Fig. 1. HOCT showed pathologic features of both renal oncocytoma and chromophobe renal cell carcinoma: for example, a tumor with a predominant architectural appearance of renal oncocytoma but with nuclear features suggestive of chromophobe renal cell carcinoma (Fig. 1b, inset). Rarely, tumor foci showed an abrupt transition between traditional-appearing renal oncocytoma-like and chromophobe renal cell carcinoma-like zones (Fig. 1c).

Pathological features of hybrid oncocytic/chromophobe renal tumors. a Gross image of a solitary hybrid oncocytic/chromophobe renal tumor arising from the superior pole of the kidney and extending into the perinephric adipose tissue. b Hybrid oncocytic/chromophobe renal tumor showing predominantly renal oncocytoma-type architecture at low power (H&E, × 4) with higher power demonstrating chromophobe renal cell carcinoma-like cytological features (inset; H&E, × 40) in the form of nuclear irregularity, atypia, and perinuclear halos. c Hybrid oncocytic/chromophobe renal tumor showing abrupt transition from a renal oncocytoma-appearing area to a chromophobe renal cell carcinoma-appearing area (H&E, × 10)
HOCTs are characterized by a low mutational frequency and a lack of mutations in renal cancer driver genes
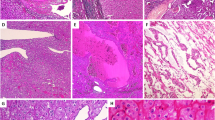
Targeted exon sequencing was performed on 16 HOCT samples derived from 14 patients. The results showed a mutational landscape of HOCTs that is characterized by a lack of mutations in classic renal cell carcinoma driver genes (i.e., VHL, BAP1, PBRM1, and SETD2 in clear cell renal cell carcinoma; FH and MET in papillary renal cell carcinoma; PTEN, TERT, and TP53 in chromophobe renal cell carcinoma; ERCC2 in renal oncocytoma; and SDHA, SDHB, SDHC, and SDHD in succinate dehydrogenase-deficient renal cell carcinoma) [11, 12, 19, 20]. Only five cases with one somatic mutation each were identified (Fig. 2a and Supplementary Table 3). The average depth of coverage among samples was ~ × 1000, ranging from 30 to × 12,000. In known hotspots and mutation loci, the depth of coverage was > × 300 (Supplementary Figure 1). Notably, our HOCT from a liver metastasis did not show mutations in the TP53 or PTEN genes but did show a frameshift deletion in the ATM gene. In addition, one other patient (#42) had a missense mutation in the ATM gene, currently classified as variant of uncertain significance. In six HOCTs cases where a suspicion of Birt–Hogg–Dubé syndrome existed, the FLCN gene was sequenced with one case that was positive for a pathogenic germline FLCN mutation (Table 2). Using publicly available data for comparison [11, 12, 20], the mutational spectrum of HOCTs is significantly different from that of chromophobe renal cell carcinoma in terms of: TP53 mutations (P = 0.005, Fisher’s exact test, Power = 0.99); mutations of either TP53 or PTEN (P = 0.0002, Fisher’s exact test, Power = 0.99); and mutations of either TP53 or PTEN or MLL3 (P = 0.000008, Fisher’s exact test, Power = 1). Despite the absence of ERCC2 mutations in HOCTs, our sample size was underpowered to demonstrate significant differences in mutations involving this gene between renal oncocytoma and HOCT.

Hybrid oncocytic/chromophobe renal tumors display low mutational frequency and limited DNA copy number alterations. a Somatic mutations in hybrid oncocytic/chromophobe renal tumor samples, showing the four mutated genes and lack of mutations in the commonly mutated renal cell carcinoma driver genes. b Copy number alterations in hybrid oncocytic/chromophobe renal tumor. Red corresponds to a gain, light blue corresponds to a partial loss of a copy of one chromosomal arm, blue corresponds to complete loss of a chromosomal arm, and pink on chromosome Y indicates female sex. Each column represents the same sample for a and b with bold borders representing two samples from the same patient. c Copy number log-ratio (logR) and B allele frequency (BAF) plots obtained by targeted sequencing analysis of tumor and normal pairs from a patient with losses on chromosomes 1 and Y and a patient without large-scale copy number alterations d
DNA copy number alteration profile of HOCTs is similar to that of renal oncocytoma
The T200.1 panel is optimized to assess whole-genome copy number alterations on the basis of coverage. Using an in-house pipeline, we found that 9 of the15 (60%) evaluable HOCT samples harbored chromosomal copy number alterations. Among these nine samples with copy number alterations, three samples showed loss only on chromosome 1 and one sample showed chromosome 1 and Y deletion. The remaining six HOCT samples (40%) showed an absence of arm-level chromosomal gains or losses. Together, the large majority of HOCT samples (67%, 10/15) showed a copy number alteration profile more similar to renal oncocytoma rather than the genome wide chromosomal copy number alterations that characterize chromophobe renal cell carcinoma (Fig. 2b). Representative plots showing HOCTs, with and without arm-spanning copy number alterations, are illustrated in Fig. 2c, d. Significantly, the B allele frequency plots showed that HOCTs are negative for the imbalanced chromosome duplication that was recently described in chromophobe renal cell carcinoma and enriched in aggressive chromophobe renal cell carcinoma [19]. The HOCT profiled from a liver metastasis did show multiple copy number alterations but its pattern of copy number losses (2p-; 5p-; 8p-; 9p-; 19p-) differs markedly from the described copy number losses in chromophobe renal cell carcinoma which typically occur in chromosomes 1, 2, 6, 10, 13, 17, 21 [11]. The metastatic HOCT did, however, show a ploidy value of 5.2, with a whole-genome doubling ratio of 1. In contrast, the non-metastatic HOCTs showed near diploid ploidy values (mean: 2.3; median: 2.2) and substantially lower whole-genome doubling ratios (mean: 0.31; median: 0.26), Supplementary Figure 2.
Expression profile of HOCTs is intermediate between that of renal oncocytoma and chromophobe renal cell carcinoma
RNA transcript data on differentially expressed genes between renal oncocytoma and chromophobe renal cell carcinoma revealed that HOCTs generally had leading fold change values that were intermediate between those of renal oncocytoma and chromophobe renal cell carcinoma (Fig. 3a and Supplementary Figure 3); a subset of genes were significantly differentially expressed in both the HOCT vs. renal oncocytoma and HOCT vs. chromophobe renal cell carcinoma contrasts (n = 24 genes with adjusted p < 0.05). However, a greater contrast was noted between HOCT vs. chromophobe renal cell carcinoma (n = 58 genes, P < 0.01) compared with HOCT vs. renal oncocytoma (n = 25 genes, P < 0.01), as illustrated in Figs. 3b, c. Notably, the HOCT cases with adverse pathologic features did not show clustering toward chromophobe renal cell carcinoma (Fig. 3a).

The gene expression profile of hybrid oncocytic/chromophobe renal tumors is intermediate between that of renal oncocytoma and chromophobe renal cell carcinoma. a Principal component analysis (PCA) of gene expression for hybrid oncocytic/chromophobe renal tumors, renal oncocytomas, and chromophobe renal cell carcinomas shows the three renal cell tumor subtypes as a continuum in which three separate clusters are visualized. Hybrid oncocytic/chromophobe renal tumors are intermediate between renal oncocytomas and chromophobe renal cell carcinomas. b and c Genes with significantly different expression assessed by the fold change (fc) between renal oncocytoma and hybrid oncocytic/chromophobe renal tumor b and between chromophobe renal cell carcinoma and hybrid oncocytic/chromophobe renal tumor c. Red, blue, and yellow bars indicate statistically significant differences in gene expression (p < 0.05). Gray bars indicate genes with non-significant differences in gene expression p > 0.05
Discussion
Herein, we present the most comprehensive clinicopathologic and molecular genetic study to date of HOCTs. We show that sporadic HOCTs are clinically and molecularly different from renal oncocytoma and chromophobe renal cell carcinoma. In comparison with renal oncocytomas or chromophobe renal cell carcinomas, we found that sporadic HOCTs were more frequently multifocal (24%), in addition to being associated with discrete renal oncocytomas elsewhere in the kidney. This is largely in agreement with previous multi-institutional studies wherein cases with hybrid renal oncocytoma—chromophobe renal cell carcinoma morphologic characteristics in a sporadic background generally showed a greater proclivity toward multifocality; with rates of multifocal renal tumors in separate series of 0% [13], 20% [21], 22% [1], and 29% [22]. By contrast, rates of multifocality in sporadic chromophobe renal cell carcinoma (4–6%) [1, 4] or renal oncocytoma (8%) [1, 23] are substantially lower.
Six of our 27 HOCTs cases presented with locally advanced or pT3a disease, in contrast to prior reports of locally advanced HOCTs by Waldert et al. [6] (2/16), Petersson et al. [13] (0/14), Pote et al. [14] (0/10), and Mai et al. [21] (0/5). The histologic characteristics of HOCTs among our cases were, however, similar to those found by others [1, 13, 14, 21, 22], with tumors showing mostly ambiguous morphology rather than a mosaic pattern with defined renal oncocytoma-like and chromophobe renal cell carcinoma-like zones. There was no evidence of microscopic necrosis or sarcomatoid change in HOCTs, pathologic features that have traditionally been associated with poor prognosis in chromophobe renal cell carcinoma [1, 4]. Importantly, our series contained two cases of HOCTs with aggressive pathologic characteristics, including a case that presented with metastatic disease and resulted in cancer specific mortality. To our knowledge, there has been only one other report of metastatic sporadic HOCT [10], although 2 of 10 cases with hybrid renal oncocytoma /chromophobe renal cell carcinoma morphology and available follow-up data in a series by Amin et al. [1] showed tumor recurrence. In the setting of Birt–Hogg–Dubé syndrome, at least four cases of HOCT with metastases have been reported, including one with sarcomatoid transformation and two tumor related deaths [9, 24]. Including the current series, the metastatic rate of HOCTs ranges between 2% in a sporadic setting [1, 6, 10, 13, 14, 21,22,23] and 5% in a Birt–Hogg–Dubé syndrome setting [9, 24, 25]. It must be acknowledged, however, that follow-up of HOCTs is limited and more mature clinical data are required to evaluate the late recurrences that sometimes occur in chromophobe renal cell carcinoma [1, 4].
HOCT is an emerging entity that has not yet been incorporated into the World Health Organization classification of tumors. Indeed, the classification of HOCT is controversial, with most of the participants at the International Society of Urologic Pathology conference indicating that they view them as a subset of chromophobe renal cell carcinoma [26]. The molecular characterization of a series of HOCTs was attempted by two groups, with conflicting results. Petersson et al. [13] analyzed 14 sporadic HOCTs by fluorescence in situ hybridization and Sanger sequencing and found either monosomy or polysomy in at least one of the chromosomes tested in all cases, with monosomy of chromosome 20 being the most common numerical aberration. The foregoing profile is distinct from those of both renal oncocytoma and chromophobe renal cell carcinoma. More recently, Pote et al. [14] evaluated copy number alterations in 10 sporadic HOCTs and two HOCTs associated with Birt–Hogg–Dubé syndrome using array-comparative genomic hybridization and fluorescence in-situ hybridization and found no chromosome imbalances in 7 of 12 cases (58%); 4 cases had chromosome 1 deletions akin to those seen in renal oncocytoma, and one case had trisomy 18. On the basis of these findings, in addition to the good outcome of their cohort and the apical Hale’s colloidal iron staining results, the authors suggested that HOCTs represent a variant of classic renal oncocytoma; however, they could not entirely exclude the possibility that HOCTs represent a separate renal tumor entity.
In this study, we analyzed the genetic profile of HOCTs in greater detail by examining, for the first time, the mutational and transcriptomic landscape of these neoplasms—in addition to their DNA copy number. Our analysis of copy number alterations is in agreement with that of Pote et al. [14] in that we showed ~ 40% of HOCTs lack chromosomal arm-spanning gains or losses; the predominant copy number alterations were losses at chromosome 1 or X/Y, similar to those described in renal oncocytoma cases and some (particularly eosinophilic variant) chromophobe renal cell carcinoma cases [11, 12]. Moreover, sequencing based tumor ploidy estimates showed HOCTs to mostly be in the near diploid range, notwithstanding the metastatic case that showed polyploidy with whole-genome doubling. Deep-targeted next-generation sequencing of HOCTs also did not identify mutations in genes described to be recurrently mutated in renal oncocytoma (ERCC2) or chromophobe renal cell carcinoma (TP53 and PTEN), including eosinophilic variant chromophobe renal cell carcinoma [11, 12, 20].
The mRNA signature of HOCT appears distinct: on the basis of an analysis of differentially expressed genes between renal oncocytoma and chromophobe renal cell carcinoma, HOCTs occupy an intermediate position between renal oncocytoma and chromophobe renal cell carcinoma. There is one prior report of three HOCTs associated with Birt–Hogg–Dubé syndrome that were assayed using an expression microarray platform and which showed the gene expression profile of HOCTs to differ from that of sporadic renal oncocytoma and chromophobe renal cell carcinoma [27], in agreement with the findings on our much larger and overwhelmingly sporadic HOCT cohort.
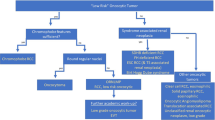
Taken together, the evidence suggests that sporadic HOCTs are clinically and pathologically distinct from renal oncocytoma and chromophobe renal cell carcinoma, and their molecular profile underscores this distinctiveness (Fig. 4). The focal chromophobe renal cell carcinoma-like cytologic features of HOCTs may suggest that they should be grouped with chromophobe renal cell carcinoma, especially eosinophilic chromophobe renal cell carcinoma, which also has few copy number alterations. However, HOCTs are more frequently multifocal, show different histopathology, lack any of the classic, recurrent mutations of chromophobe renal cell carcinoma, and differ transcriptomically from chromophobe renal cell carcinoma. HOCTs also differ genetically from renal oncocytoma and notably, they show low but definite malignant potential, unlike the uniformly benign renal oncocytoma [23]. Despite these differences, HOCTs are nevertheless more closely related to renal oncocytoma and chromophobe renal cell carcinoma than to any other renal cell carcinoma subtype at the pathological, clinical, and molecular levels. As the data mature and a larger number of cases are molecularly assayed, it is expected that HOCTs will occupy an intermediate position in the spectrum of renal oncocytic neoplasia.

Schematic illustration of the spectrum of renal oncocytic neoplasms. The figure summarizes the characteristic genomic findings among renal oncocytomas, hybrid oncocytic/chromophobe renal tumors, and chromophobe renal cell carcinomas at the copy number alteration, mutation, and gene expression levels. The bottom heatmap highlights the eight most differentially expressed genes among the three entities
Change history
09 July 2019
The original version of this article was updated to correct the author L. Jeffrey Medeiros’ name to first name: ‘L. Jeffrey’ last name: ‘Medeiros’ in HTML version.
References
Amin MB, Paner GP, Alvarado-Cabrero I, Young AN, Stricker HJ, Lyles RH, et al. Chromophobe renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 145 cases. Am J Surg Pathol. 2008;32:1822–34.
Hes O, Petersson F, Kuroda N, Hora M, Michal M. Renal hybrid oncocytic/chromophobe tumors - a review. Histol Histopathol. 2013;28:1257–64.
Kryvenko ON, Jorda M, Argani P, Epstein JI. Diagnostic approach to eosinophilic renal neoplasms. Arch Pathol Lab Med. 2014;138:1531–41.
Przybycin CG, Cronin AM, Darvishian F, Gopalan A, Al-Ahmadie HA, Fine SW, et al. Chromophobe renal cell carcinoma: a clinicopathologic study of 203 tumors in 200 patients with primary resection at a single institution. Am J Surg Pathol. 2011;35:962–70.
Sircar K, Rao P, Jonasch E, Monzon FA, Tamboli P. Contemporary approach to diagnosis and classification of renal cell carcinoma with mixed histologic features. Chin J Cancer. 2013;32:303–11.
Waldert M, Klatte T, Haitel A, Ozsoy M, Schmidbauer J, Marberger M, et al. Hybrid renal cell carcinomas containing histopathologic features of chromophobe renal cell carcinomas and oncocytomas have excellent oncologic outcomes. Eur Urol. 2010;57:661–5.
Leveridge MJ, Finelli A, Kachura JR, Evans A, Chung H, Shiff DA, et al. Outcomes of small renal mass needle core biopsy, nondiagnostic percutaneous biopsy, and the role of repeat biopsy. Eur Urol. 2011;60:578–84.
Pavlovich CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. 2002;26:1542–52.
Benusiglio PR, Giraud S, Deveaux S, Mejean A, Correas JM, Joly D, et al. French National Cancer Institute Inherited Predisposition to Kidney Cancer N: Renal cell tumour characteristics in patients with the Birt-Hogg-Dube cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis. 2014;9:163.
Aslam MI, Spencer L, Garcea G, Pollard C, Metcalfe MS, Harrison RF, et al. A case of liver metastasis from an oncocytoma with a focal area of chromophobe renal cell carcinoma: a wolf in sheep’s clothing. Int J Surg Pathol. 2009;17:158–62.
Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–30.
Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21.
Petersson F, Gatalica Z, Grossmann P, Perez Montiel MD, Alvarado Cabrero I, Bulimbasic S, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch. 2010;456:355–65.
Pote N, Vieillefond A, Couturier J, Arrufat S, Metzger I, Delongchamps NB, et al. Hybrid oncocytic/chromophobe renal cell tumours do not display genomic features of chromophobe renal cell carcinomas. Virchows Arch. 2013;462:633–8.
Chen K, Meric-Bernstam F, Zhao H, Zhang Q, Ezzeddine N, Tang LY, et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem. 2015;61:544–53.
Rohan S, Tu JJ, Kao J, Mukherjee P, Campagne F, Zhou XK, et al. Gene expression profiling separates chromophobe renal cell carcinoma from oncocytoma and identifies vesicular transport and cell junction proteins as differentially expressed genes. Clin Cancer Res. 2006;12:6937–45.
Tan MH, Wong CF, Tan HL, Yang XJ, Ditlev J, Matsuda D, et al. Genomic expression and single-nucleotide polymorphism profiling discriminates chromophobe renal cell carcinoma and oncocytoma. BMC Cancer. 2010;10:196.
Wang Z, Kim TB, Peng B, Karam JA, Creighton CJ, Joon AY, et al. Sarcomatoid renal cell carcinoma has a distinct molecular pathogenesis, driver mutation profile and transcriptional landscape. Clin Cancer Res 2017;23:6686–6696.
Casuscelli J, Weinhold N, Gundem G, Wang L, Zabor EC, Drill E, et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI Insight. 2017;2: pii: 92688.
Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 2015;13:1895–908.
Mai KT, Dhamanaskar P, Belanger E, Stinson WA. Hybrid chromophobe renal cell neoplasm. Pathol Res Pract. 2005;201:385–9.
Delongchamps NB, Galmiche L, Eiss D, Rouach Y, Vogt B, Timsit MO, et al. Hybrid tumour ‘oncocytoma-chromophobe renal cell carcinoma’ of the kidney: a report of seven sporadic cases. BJU Int. 2009;103:1381–4.
Trpkov K, Yilmaz A, Uzer D, Dishongh KM, Quick CM, Bismar TA, et al. Renal oncocytoma revisited: a clinicopathological study of 109 cases with emphasis on problematic diagnostic features. Histopathology. 2010;57:893–906.
Houweling AC, Gijezen LM, Jonker MA, van Doorn MB, Oldenburg RA, van Spaendonck-Zwarts KY, et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105:1912–9.
Pavlovich CP, Grubb RL 3rd, Hurley K, Glenn GM, Toro J, Schmidt LS, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol. 2005;173:1482–6.
Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, et al. The International Society of Urological Pathology (ISUP) vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89.
Klomp JA, Petillo D, Niemi NM, Dykema KJ, Chen J, Yang XJ, et al. Birt-Hogg-Dube renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression. BMC Med Genomics. 2010;3:59.
Acknowledgements
This work was supported by an Intradepartmental Research Grant from the Department of Pathology at The University of Texas MD Anderson Cancer Center to RRC, and the Kidney Cancer Research Program (Monteleone Foundation) to KS. We acknowledge Kim-Anh Vu and Ann Sutton for technical and secretarial assistance. Exome-seq data were generated by the UT-MDACC Sequencing and Microarray Facility (NCI Core grant CA016672 SMF). Some work was supported in part with support from the Khalifa Foundation and the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ruiz-Cordero, R., Rao, P., Li, L. et al. Hybrid oncocytic/chromophobe renal tumors are molecularly distinct from oncocytoma and chromophobe renal cell carcinoma. Mod Pathol 32, 1698–1707 (2019). https://doi.org/10.1038/s41379-019-0304-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0304-y
This article is cited by
-
Convolutional neural networks for the differentiation between benign and malignant renal tumors with a multicenter international computed tomography dataset
Insights into Imaging (2024)
-
Intraoperative MRI-derived volumetric ablation margins and initial correlation with local outcome after MRI-guided cryoablation of renal tumors
Cancer Imaging (2023)
-
A pilot radiometabolomics integration study for the characterization of renal oncocytic neoplasia
Scientific Reports (2023)
-
Do we need an updated classification of oncocytic renal tumors?
Modern Pathology (2022)
-
Onkozytäre Tumoren der Niere – neue Differenzialdiagnosen
Der Pathologe (2021)

{kind=link}